UCSD Researchers Use AMD GPUs to Build the World’s Largest SARS-CoV-2 Alignment and Pangenome
Feb 17, 2026

A research team at the University of California San Diego (UCSD), led by Prof. Yatish Turakhia, is pioneering new GPU-accelerated approaches to tackle some of the most fundamental and computationally intensive problems in bioinformatics, such as constructing large multiple-sequence alignments and pangenomes. A multiple sequence alignment (MSA) arranges genomic sequences as rows in a matrix such that homologous positions, those inferred to have a shared evolutionary origin, are aligned in the same columns, allowing for the identification of genomic positions that have acquired mutations across genomes. A pangenome represents the complete collection of genomes from a single species that captures the full spectrum of genetic variation present across its population. Together, MSAs and pangenomics provide a critical foundation for studying genome evolution and understanding how mutations influence biological traits of a species, such as increased virulence, transmissibility, or drug resistance in a pathogen.
Despite the abundance of genomic data for many species, constructing their MSAs and pangenomes at scale remains a formidable challenge. For instance, SARS-CoV-2, the pathogen responsible for COVID-19, has over 16 million genomes available in the GISAID database, yet until recently, no comprehensive pangenome was available. Conventional MSA tools struggled at this scale, often relying on reference-based shortcuts that speed up the alignment process but compromise on accuracy. To overcome this barrier, the UCSD team developed a GPU-accelerated tool for ultra-fast inference of MSA and an information-rich, highly compressible pangenome format to make population-scale pangenomic analysis feasible for the first time.
Ultra-large Multiple Sequence Alignments
This challenge of inferring population-scale MSAs was addressed by Yu-Hsiang Tseng, a computer engineering PhD student in Turakhia’s lab. Yu-Hsiang developed TWILIGHT, a GPU-accelerated tool that is uniquely capable of efficiently aligning massive numbers of extremely long sequences. TWILIGHT was published last year in Bioinformatics as part of the proceedings for the flagship computational biology conference, Intelligent Systems for Molecular Biology (ISMB). TWILIGHT combines a divide-and-conquer alignment strategy with memory-efficient data structures, enabling the tool to compute ultra-large MSAs within the limited GPU memory such as MAFFT. This design reduces runtimes from days to minutes compared to state-of-the-art CPU-based tools. As shown in Figure 1, TWILIGHT completes the alignment in 257 seconds on an AMD Instinct™ MI210 GPU (56x faster than MAFFT on a 32-core CPU) and in just 177 seconds on an AMD Instinct MI300X GPU (82x faster).
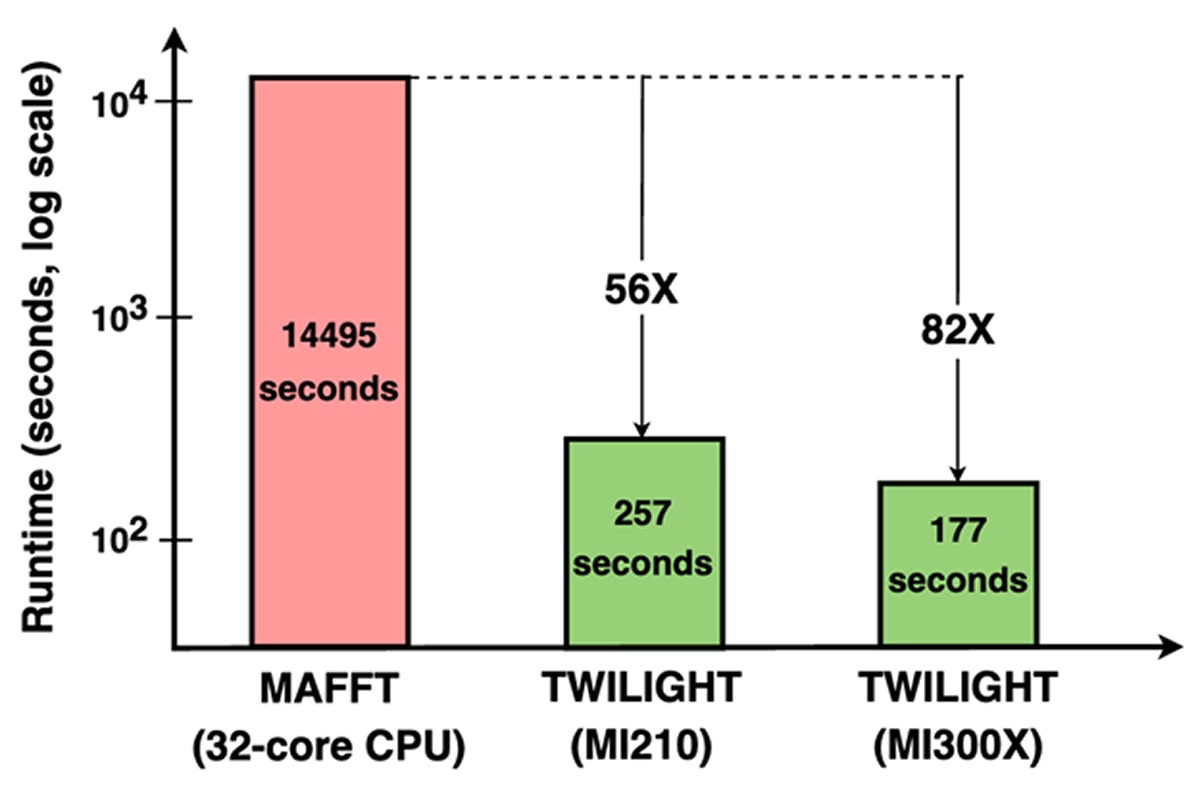
Leveraging this capability, TWILIGHT has now produced the first reference-free SARS-CoV-2 MSA covering 16.2 million genomes, excluding only low-quality sequences, completing the alignment in 48 hours on a single server node powered by four AMD Instinct MI210 GPUs and two 64-core AMD EPYC™ processors, demonstrating the practicality of population-scale MSA construction on modern GPU-accelerated systems.
"A reference-free MSA at this scale allows researchers to finally study the full spectrum of virus’ mutations, something that was not possible with previous subsampling or reference-based approaches," said Yu-Hsiang Tseng. “This work was greatly accelerated using AMD GPUs.”
Compressive Pangenomics
The SARS-CoV-2 MSA has also enabled the UCSD team to construct the largest SARS-CoV-2 pangenome to date using a new approach they developed called “compressive pangenomics”. While existing pangenomic data formats struggle to scale to millions of genomes and primarily capture variation across genome collections, the team introduced a novel data structure and file format, called the Pangenome Mutation-Annotated Network (PanMAN)ss, which preserves phylogeny, mutational history, and whole-genome alignment while achieving extreme compression, enabling analysis at unprecedented scale.
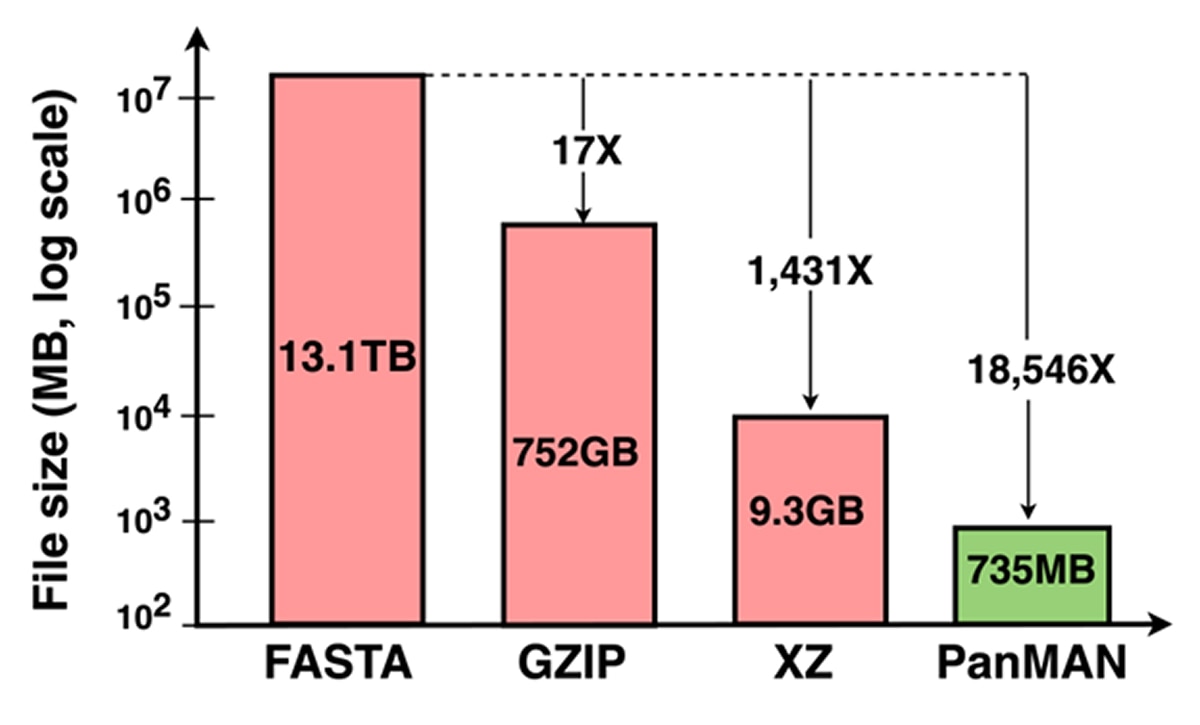
As shown in Figure 2, PanMAN even outperforms traditional compression methods, such as GZIP, XZ, and AGC, which only store raw genomic sequences without any additional biological inferences derived from them. The team recently published this compressive pangenomics technique in Nature Genetics and released online a SARS-CoV-2 pangenome of 8 million unrestricted genome sequences. This PanMAN requires only 366MB of file storage space, which is roughly 3,000x smaller than the whole-genome alignment that it encodes. Building on this success, the team has now constructed an even larger pangenome from 16.2 million user-restricted GISAID sequences using the TWILIGHT-based MSA and UShER-based phylogeny, which is maintained by collaborators at UC Santa Cruz, Prof. Russell Corbett-Detig and Angie Hinrichs. This pangenome, requiring only 735 MB of disk space, is likely the largest SARS-CoV-2 pangenome to date, and is available on request to registered GISAID users.
“What makes this result exciting isn’t just the unprecedented scale, but what it enables, scientifically,” said Sumit Walia, a computer engineering Ph.D. candidate in Turakhia’s lab and the lead developer of PanMAN. “Our representation offers a more detailed portrayal of the pathogen's evolutionary and mutational history than anything previously available, facilitating the discovery of new biological insights.”
Looking Forward: Ultra-large Human Pangenomes
The team is now extending TWILIGHT and PanMAN from viral and microbial datasets to the more complex space of human pangenomics. This work is also being supported by AMD. This extension could fundamentally transform the scale and speed at which human genetic data are stored, analyzed, and shared, and deepen our understanding of how the genome influences human evolution and disease.
"It will be exciting to see how the remarkable compression results achieved by PanMAN on microbial datasets carry over to human pangenomes," said Benedict Paten, Professor at UC Santa Cruz and a co-leader of the Human Pangenome Reference Consortium (HPRC). “Scaling pangenomics to large human populations has long been constrained by computational and memory barriers, which this approach can substantially ease.”
“We are thrilled to partner with AMD on this journey,” said Yatish Turakhia. “AMD’s resources have allowed us to perform research at scales that would be difficult to imagine otherwise.”
TWILIGHT: https://github.com/TurakhiaLab/TWILIGHT
PanMAN: https://github.com/TurakhiaLab/panman

Contributors

Related Blogs
-
Understanding Attention Algorithms and Their Backends for Image and Video Generation — ROCm Blogs
Practical guide to attention backends in ComfyUI on AMD describing how to optimize performance, memory, and stability with the right configuration.
July 19, 2026
-
Efficient Hyperparameter Optimization for Autonomous Driving Models with AMD Instinct GPU Partitioning — ROCm Blogs
Accelerate HPO for autonomous driving models using AMD MI300X GPU partitioning for higher throughput, efficiency, and parallelism.
July 07, 2026
-
A Practical Guide to Running LLMs on AMD Radeon™ GPUs — ROCm Blogs
This guide describes how to run LLMs on AMD Radeon™ GPUs using a range of partner frameworks, tools, and runtimes, with step-by-step setup instructions and performance optimization tips.
June 18, 2026
-
AI Across Industries: How AMD Technologies Are Powering the Next Wave of Enterprise Transformation
AI is reshaping every industry. Explore how leading enterprises are using AMD-powered AI infrastructure to accelerate innovation, improve efficiency, and drive business transformation.
June 18, 2026
-
Productionizing TurboQuant on AMD GPUs for KV-Cache-Bound LLM Inference — ROCm Blogs
Productionized TurboQuant 4-bit KV-cache quantization on AMD GPUs via vLLM, with custom kernels and accuracy analysis on agentic workloads.
June 10, 2026
-
AMD-Powered 3D Gaussian Splatting for Autonomous Driving Scenes — ROCm Blogs
Run Street Gaussians on AMD Instinct MI300: migrate to latest gsplat, install on ROCm, and render dynamic street scenes.
May 06, 2026
-
Deploy and Customize AMD Solution Blueprints — ROCm Blogs
Learn how to deploy and customize AMD Solution Blueprints — from default deployment to swapping and reusing AMD Inference Microservices across multiple blueprints.
April 01, 2026
-
Solution Blueprints: Accelerating AI Deployment with AMD Enterprise AI — ROCm Blogs
This blog presents AIMs Solution Blueprints and demonstrates modular, Helm‑based deployment patterns.
February 10, 2026







